Algorithm and format requirements
Algorithm Format requirements
Algorithm & format requirements
Help
Contact us
Many protein families contain homologous proteins that have a common biological function, but different specificity towards substrates, ligands, effectors, DNA, proteins and other interacting molecules including other monomers of the same protein. All these interactions must be highly specific. Our aim is to find groups of protein with same specificity (specificity group) and amino acid residues, that determine this specificity.
Amino acid residues that determine differences in protein functional specificity and account for correct recognition of interaction partners, are usually thought to correspond to those positions of a protein multiple alignment, where the distribution of amino acids is closely associated with grouping of proteins by specificity. SDPfox searches for division into specificity groups and positions that are well conserved within this groups but differ between them. These positions are called SDPs (specificity-determining positions).
SDPfox includes the following interconnected procedures: SDPlight to predict SDPs, SDPprofile to assign specificity to unannotated proteins, SDPgroup to split family into groups of specificity from a training sample (a small number of proteins from the considered family, for which specificity is known), SDPclust to construct a cluster tree of protein specificity. SDPlight, SDPprofile and SDPgroup is available at web-server SDPfox, all methods of SDPfox is realized as a stand-alone console program SDPfox.
Consider a multiple protein sequence alignment. The proteins are divided into N specificity
groups, numbered by i=1,...,N. The goal in to identify columns (positions) in the alignment,
in which the amino acid distribution is closely associated with the grouping by specificity.
This association in column p of the alignment is measured by the mutual information
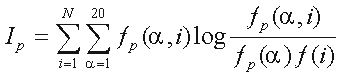 ,
,
where 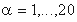 is a residue type,
is a residue type, 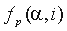 is the ratio of the number of occurrences of residue
is the ratio of the number of occurrences of residue  in group i at position p to the length of the whole alignment column,
in group i at position p to the length of the whole alignment column,
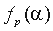 is the frequency of residue
in the whole alignment column,
is the frequency of residue
in the whole alignment column, 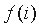 is the fraction of proteins belonging
to group i. The mutual information reflects the statistical association between two discrete random variables
and i.
is the fraction of proteins belonging
to group i. The mutual information reflects the statistical association between two discrete random variables
and i.
To address the facts that frequencies are calculated based on a small sample, and that substitutions to amino
acids with similar physical properties should be weakly penalized, the observed amino acid frequencies are modified.
Instead of using 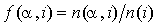 , where
, where 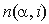 is the number of occurrences of residue in group i,
is the number of occurrences of residue in group i,
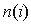 is the size of group i (here i is a single group or
the whole alignment), SDPlight uses smoothed frequencies
is the size of group i (here i is a single group or
the whole alignment), SDPlight uses smoothed frequencies
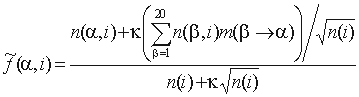 ,
,
where 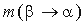 is the probability of amino acid substitution
is the probability of amino acid substitution
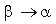 according to the matrix corresponding to the average identity in group
i,
according to the matrix corresponding to the average identity in group
i, 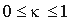 is a smoothing parameter.
is a smoothing parameter.
To calculate the statistical significance of the obtained values of
Ip, average of distribution and dispersion of mutual information (M(Ip)
and D(Ip)) are calculated from theoretical knowledge. To offset the background similarity of proteins
that is higher within groups than between groups, we calculate the expected mutual information for the column p
Iexp=aM(I)+b where a and b do not depend on the position, i.e.
are the same for every position of the alignment , so that
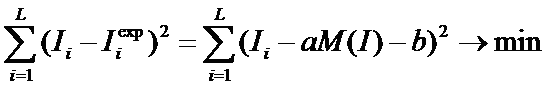
L is the total length of the alignment, 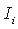 is the observed mutual information for the i-th column.
is the observed mutual information for the i-th column.
Then, Z-scores are calculated:
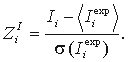
A high value of Z-scores indicates a position, where the amino acid distribution is much closer associated with
grouping by specificity than for an average position of the alignment, and thus, which is likely to be an SDP.
Given a series of Z-scores corresponding to every position
of the multiple alignment, one needs to evaluate the significance of the Z-scores in order to tell whether the
observed Z-score is sufficiently high to indicate a SDP. SDPpred uses an automated procedure for setting the
thresholds based on the computation of the Bernoulli estimator. The observed Z-scores are oredered by
decrease: 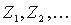 . The threshold is defined as:
. The threshold is defined as:
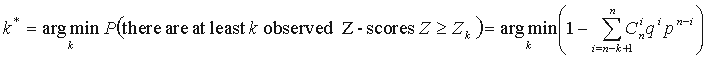
where n is the total number of considered positions, 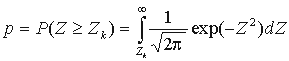 ,
,
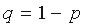 .
. 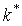 positions having
highest Z-scores are designated SDPs, as they are the least probable to constitute a tail of the Gaussian
distribution, and thus are non-randomly generated positions. p(k*) is
further referred as p-value.
positions having
highest Z-scores are designated SDPs, as they are the least probable to constitute a tail of the Gaussian
distribution, and thus are non-randomly generated positions. p(k*) is
further referred as p-value.
To predict specificity of other proteins, we calculate a profile matrix based on SDPs for each group:
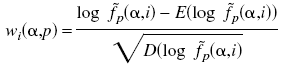
is weight of amino acids in the position p in the profile for group i.
Then, for each protein we calculate profile weights for all specificity groups: 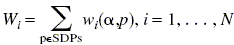 ,
,
and select the group providing the maximal weight. Then one can assume that the considered protein has a specificity coinciding with the specificity of the maximal weight.
SDPgroup is an iterative procedure for splitting family into groups of specificity using training sample:
- Initiation: proteins from the training sample form initial specificity groups.
- Step of iteration: SDPs are identified with SDPlight. For all sequences of the family, profile scores for all specificity groups are calculated using SDPprofile. Sequences are rearranged according to the maximal weight
- End: The step of iteration does not result in rearramgement of sequences
SDPclust is a stochastic procedure to construct specificity cluster tree:
- The family is randomly split into a large number of specificity groups
- Starting from this splitting as a training sample, SDPgroup procedure is performed
- Steps 1-2 were repeated 10000 times. A cluster tree was built based on how often two sequences fall into same specificity group.
- The specificity groups are extracted from the tree so that the weight of worst group is the highest among
all possible clustering that emerge from tree. The weight Cw is defined as follows:
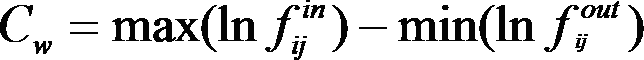
where fijin and fijout - are frequencies of grouping of sequences i and j together or separately, respectively, max is taken for all i and j from current cluster, min is taken for all i from current cluster and all j outside of current cluster, for cluster, which contain only one sequence weight is accepted equal zero.
The only information needed for prediction of SDPs is a multiple alignment of protein sequences divided into
specificity groups. The aligned sequences should be in the GDE or fasta format. Amino acids should be indicated by
small or big characters. Gaps should be indicated by dots or hyphen. Name of each sequence is placed onto a
separate line and begins with '%' or '>'.
The alignment may be manually edited in order to define the specificity groups. They may be
separated by lines beginning and ending with the "equals" sign and containing name of the following group, e.g.:
"===Group1===".
The alignment should contains from 4 to 2000 sequences, not more than 200 groups and be shorter that 5000 aa.
Thus the input alignment should look like this: