SDPpred: a tool for prediction of amino acid residues that determine differences in functional specificity of homologous proteins |
Home & Submit job |
Algorithm & |
Contact us |
SDPpred is a tool for prediction of residues in protein sequences that determine functional differences between proteins, having same general biochemical function.
Many protein families contain homologous proteins that have a common biological function, but different specificity towards substrates, ligands, effectors, DNA, proteins and other interacting molecules including other monomers of the same protein. All these interactions must be highly specific. Our aim is to find amino acid residues, which account for different specificity of proteins from one family, i.e. to distinguish amino acid substitutions caused by random evolutionary process from those caused by switch of specificity.
Amino acid residues that determine differences in protein functional specificity and account for correct recognition of interaction partners, are usually thought to correspond to those positions of a protein multiple alignment, where the distribution of amino acids is closely associated with grouping of proteins by specificity. SDPpred searches for positions that are well conserved within specificity groups but differ between them. These positions are called SDPs (specificity-determining positions). Such positions, though obvious in alignments containing a small number of proteins and specificity groups, become a challenge to find in large protein families with a variety of specificities.
The only information required for prediction of SDPs is a multiple alignment of protein sequences divided into specificity groups (more details on the input format here). SDPpred can analyze alignments of length up to 2000 positions, containing at most 1000 proteins. There can be up to 1000 specificity groups. However, it is recommended that each group would contain at least three sufficiently divergent sequences. On the other hand, the average identity in each group should not be less than 25%. Having more than two groups also strongly improves the quality of prediction due to more efficient elimination of the background evolutionary similarity.
SDPpred predicts a set of SDPs, maps them onto the multiple alignment of the protein family or onto a user-selected protein in this alignment (more detail on the output format here).
SDPpred
- Does not use any information about the proteins' structure. The procedure is based solely on statistical analysis of an alignment, and thus it can be applied to protein families that do not include any members with resolved 3D structure. Automatically calculates the number of SDPs and the probability of occurrence of these positions by chance (B-cutoff setting). It does not use any ad hoc cutoff and thus does not require any prior knowledge about special properties of the analyzed family. Substitutions within specificity groups are weighted according to physical properties of amino acids, using a substitution matrix, so that substitutions to amino acids with similar properties are only weakly penalized.
- Incorporates information about evolutionary distance within and between groups by using different amino acid substitution matrices.
Algorithm
Consider a multiple protein sequence alignment. The proteins are divided into N specificity groups, numbered by i=1,...,N. The goal in to identify columns (positions) in the alignment, in which the amino acid distribution is closely associated with the grouping by specificity. This association in column p of the alignment is measured by the mutual information
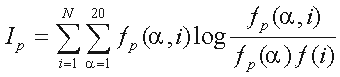 ,
,
where 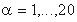 is a residue type,
is a residue type, 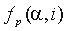 is the ratio of the number of occurrences of residue
is the ratio of the number of occurrences of residue 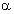 in group i at position p to the length of the whole alignment column,
in group i at position p to the length of the whole alignment column, 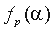 is the frequency of residue in the whole alignment column,
is the frequency of residue in the whole alignment column, 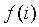 is the fraction of proteins belonging to group i. The mutual information reflects the statistical association between two discrete random variables and i.
is the fraction of proteins belonging to group i. The mutual information reflects the statistical association between two discrete random variables and i.
To address the facts that frequencies are calculated based on a small sample, and that substitutions to amino acids with similar physical properties should be weakly penalized, the observed amino acid frequencies are modified. Instead of using 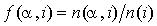 , where
, where 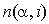 is the number of occurrences of residue in group i,
is the number of occurrences of residue in group i, 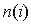 is the size of group i (here i is a single group or the whole alignment), SDPpred uses smoothed frequencies
is the size of group i (here i is a single group or the whole alignment), SDPpred uses smoothed frequencies
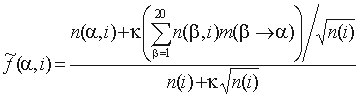 ,
,
where 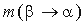 is the probability of amino acid substitution
is the probability of amino acid substitution 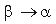 according to the matrix corresponding to the average identity in group i,
according to the matrix corresponding to the average identity in group i, 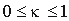 is a smoothing parameter.
is a smoothing parameter.
To calculate the statistical significance of the obtained values of Ip, each column is shuffled, yielding the distribution 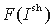 . To offset the background similarity of proteins that is higher within groups than between groups, we calculate the expected mutual information for the column p
. To offset the background similarity of proteins that is higher within groups than between groups, we calculate the expected mutual information for the column p 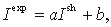 where a and b do not depend on the position, i.e. are the same for every position of the alignment , so that
where a and b do not depend on the position, i.e. are the same for every position of the alignment , so that
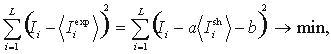
L is the total length of the alignment, 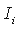 is the observed mutual information for the i-th column.
is the observed mutual information for the i-th column.
Then, Z-scores are calculated:
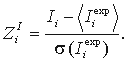
A high value of Z-scores indicates a position, where the amino acid distribution is much closer associated with grouping by specificity than for an average position of the alignment, and thus, which is likely to be an SDP.
Given a series of Z-scores corresponding to every position of the multiple alignment, one needs to evaluate the significance of the Z-scores in order to tell whether the observed Z-score is sufficiently high to indicate a SDP. SDPpred uses an automated procedure for setting the thresholds based on the computation of the Bernoulli estimator. The observed Z-scores are oredered by decrease: 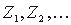 . The threshold is defined as:
. The threshold is defined as:
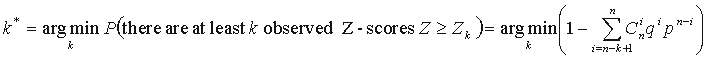
where n is the total number of considered positions, 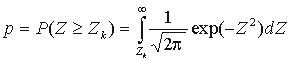 ,
, 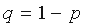 .
. 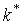 positions having highest Z-scores are designated SDPs, as they are the least probable to constitute a tail of the Gaussian distribution, and thus are non-randomly generated positions. p(k*) is further referred as p-value.
positions having highest Z-scores are designated SDPs, as they are the least probable to constitute a tail of the Gaussian distribution, and thus are non-randomly generated positions. p(k*) is further referred as p-value.
To predict specificity of other proteins, we calculate a profile matrix based on SDPs for each group:
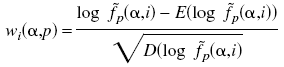
is weight of amino acids in the position p in the profile for group i.
Then, for each new protein we calculate profile weights for all specificity groups: 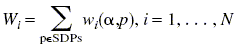 ,
,
and select the group providing the maximal weight. Then one can assume that the considered protein has a specificity coinciding with the specificity of the maximal weight. To estimate statistical significance of this observation, we calculate z-scores:
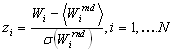
High z-score incombination with high profile weight indicate that the considered protein has the assumed specificity.
Input and output format
Input
The only information needed for prediction of SDPs is a multiple alignment of protein sequences divided into specificity groups. The aligned sequences should be in the GDE format. Columns containing small characters or dots ('.') will be ignored. The alignment should be manually edited in order to define the specificity groups. They should be separated by lines beginning with the "equals" sign and containing name of the following group, e.g.
=Group1
Generally, the group name can be framed by any number of spaces and the "equals" signs, e.g. '=== Group1 ===' is also a valid header for the group named 'Group1'.
Thus the input alignment should look like this:
On a latter stage, the user should also select the number of shuffles for computation of the statistical significance (between 1 000 and 10 000). An alignment of a thousand of sequences divided into several hundreds of specificity groups is analyzed in a couple of hours if each column is shuffled 10 000 times. Using less shuffles reduces the required time proportionally, but makes the results less reliable. Typically, the top of the SDP list remains the same, but minor variations may appear near the cutoff.
It is also possible to improve the prediction by predicting transmembrane (TM) segments of the proteins first (using forward-backward algorithm with TMHMM-like hidden markov model for multiple alignment profile, followed by filtering columns with high posterior TM-probability) and then using special TM matrices for the TM segments (described here).
Output
SDPpred outputs the set of SDPs, i.e. positions of the alignment, which are likely to determine differences in functional specificity between the provided groups. These positions exhibit amino acid distribution highly correlated with grouping by specificity.
The set of SDPs can be visualized in several ways:
- As the list of predicted SDPs. The probability of obtaining of a given number of SDPs by chance is given in the second right column of the table. By clicking on 'Details' the user cansee the distribution of amino acids for the given SDPs ina separate window.
- In a separate window, the colored alignment can be displayed.
- Below the list of SDPs, all local minima of p-value are displayed. Each local minimum can be selected as threshold of the list of SDPs. The first two display options will be modified accordingly. It is reasonable to consider different local minima if they have close p-values to the global minimum. In this case, some alterations due to the randomization procedure can occur between runs.
- A link from the results page leads to specificity prediction window. On this page, the user can load a number of sequences, alighned to the initial alignment, to predict their specificity. Only specificities coinciding with specificity of one of initial groups are considered. As the result of this prediction, the user gets a table containing profile weights for all specificity groups, maximal profile weight, name of the group providing this maximal weight, maximal z-score and the name of the group providing the latter. In additional window, amino acid residues occupying SDPs in the query proteins can be displayed.